
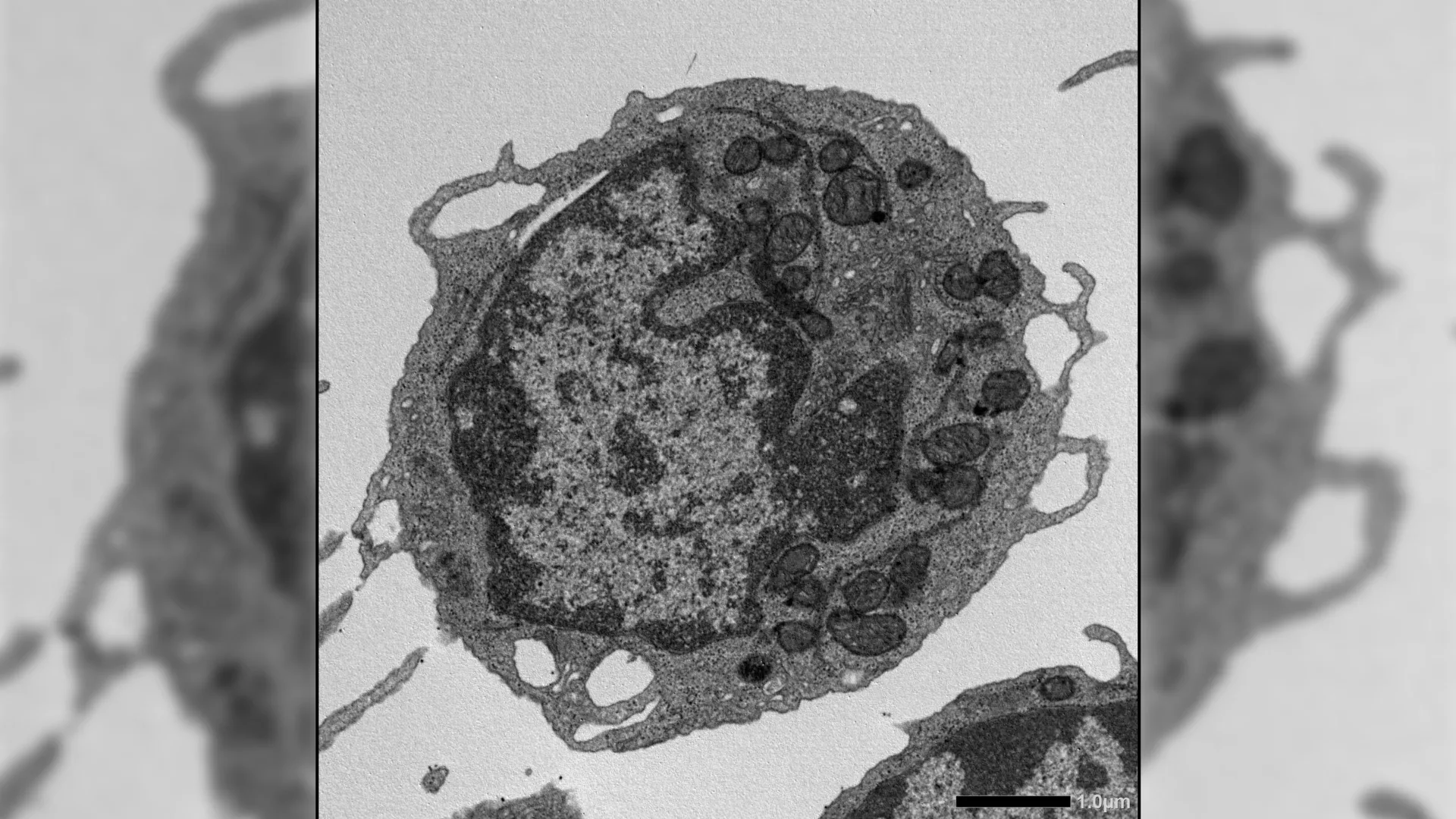
As organisms advance in age, a discernible decline in the robustness of the circulatory and immune systems becomes increasingly apparent, a phenomenon largely attributable to the diminishing capacity of hematopoietic stem cells (HSCs). These foundational cells, residing within the bone marrow, are tasked with the critical responsibility of generating all blood cell lineages, a process that must be meticulously balanced throughout life. Under optimal conditions, HSCs possess a remarkable ability to self-renew, thereby maintaining a diverse and functional pool of blood components. However, with the passage of time, this regenerative efficiency wanes significantly. The production of new cells falters, a skewed output emerges favoring certain cell types like myeloid progenitors at the expense of others, such as lymphoid cells, and consequently, the capacity to mount an effective immune defense is compromised.
The multifaceted nature of this age-related deterioration in HSC function is influenced by a constellation of factors. These include the cumulative burden of cellular damage accrued over a lifetime, subtle yet persistent alterations in gene expression patterns, a chronic state of low-grade inflammation permeating the body, and shifts in the microenvironment of the bone marrow, which serves as the stem cells’ niche. Despite a growing understanding of these individual stressors, the intricate interplay between these diverse challenges and their collective impact on impairing HSC efficacy has remained an area of considerable scientific inquiry.
A collaborative endeavor involving researchers from The University of Tokyo, Japan, and St. Jude Children’s Research Hospital, USA, has shed new light on this complex biological process. Their investigation delved into the specific mechanisms by which age-associated stressors exert their influence on HSCs, with a particular focus on a signaling pathway known as the receptor-interacting protein kinase 3 (RIPK3)-mixed lineage kinase like (MLKL) axis. This pathway has historically been primarily associated with necroptosis, a distinct form of programmed cell death, prompting the researchers to explore its potential role beyond its established function.
Spearheading this pivotal research was Dr. Masayuki Yamashita, who at the time of the study held positions as an Assistant Member at St. Jude Children’s Research Hospital and an Assistant Professor at The Institute of Medical Science, The University of Tokyo. Key contributions to the study were also made by Dr. Atsushi Iwama from The Institute of Medical Science, The University of Tokyo, and Dr. Yuta Yamada, a graduate student at The Institute of Medical Science, The University of Tokyo, who conducted their research at St. Jude Children’s Research Hospital.
An unexpected observation initiated the research trajectory, leading to a paradigm-shifting insight into the function of MLKL. Dr. Yamashita articulated this pivotal moment, stating, "We discovered an unexpected phenotype in HSCs of MLKL-knockout mice repeatedly treated with 5-fluorouracil, where aging-associated functional changes were markedly attenuated despite no detectable difference in HSC death, prompting us to investigate whether this pathway might induce functional changes beyond cell death." This remarkable finding suggested that MLKL could influence the aging process of stem cells without necessarily leading to their demise, a hypothesis that became the central tenet of their subsequent investigations. The comprehensive findings of this groundbreaking research were formally published in Volume 17 of Nature Communications on April 6, 2026.
To rigorously test this novel hypothesis, the research team employed a sophisticated array of experimental models. This included the utilization of genetically modified mice, specifically those engineered to be deficient in either MLKL or RIPK3, alongside wild-type counterparts for comparative analysis. Furthermore, specialized reporter mice were developed, incorporating a Förster resonance energy transfer (FRET)-based biosensor, which enabled the real-time detection and quantification of MLKL activation within the cells. These carefully designed models allowed the researchers to meticulously dissect the role of the RIPK3-MLKL axis in the context of aging.
The experimental protocols involved exposing these mouse models to a series of controlled stress conditions meticulously chosen to recapitulate the various insults experienced by cells during the aging process. These included inducing states of inflammation, replication stress, and oncogenic stress, thereby simulating key drivers of cellular senescence and dysfunction. The functional integrity of HSCs was primarily assessed through bone marrow transplantation assays, a gold standard technique that evaluates the capacity of transplanted stem cells to reconstitute the entire blood system, a direct measure of their regenerative potential.
Complementing these in vivo assessments, a battery of advanced molecular and cellular techniques provided granular insights into the underlying mechanisms. These included flow cytometry for cell surface marker analysis and cell sorting, ex vivo expansion assays to assess proliferative capacity, RNA sequencing (RNA-seq) to comprehensively map gene expression profiles, assay for transposase-accessible chromatin sequencing (ATAC-seq) to probe chromatin accessibility and regulatory regions, high-resolution imaging for detailed morphological analysis, metabolic testing to evaluate cellular energy production, and detailed studies of mitochondrial function. The synergistic application of these diverse methodologies enabled the researchers to scrutinize the impact of MLKL on HSCs at multiple, interconnected levels of cellular organization and function.
The experimental results yielded a profound and previously unrecognized function for MLKL in the intricate process of stem cell aging. Contrary to its established role in inducing cell death, the activation of MLKL within HSCs under stress conditions did not correlate with an increased rate of cell demise or a reduction in overall cell numbers. Instead, its influence manifested through a distinctly different mechanism.
Upon activation in response to cellular stress, MLKL was observed to transiently translocate to the mitochondria, the powerhouses of the cell responsible for energy generation. Within these vital organelles, MLKL instigated damage by disrupting the mitochondrial membrane potential, altering the structural integrity of the mitochondria, and consequently impairing their energy-producing capabilities. These detrimental effects on mitochondrial function were directly linked to the hallmarks of HSC aging, including a diminished capacity for self-renewal, a reduced output of lymphoid progenitor cells, and a pronounced skewing of differentiation towards the myeloid lineage.
Crucially, when MLKL was genetically ablated or pharmacologically inhibited, a significant amelioration of these age-related defects was observed. HSCs lacking functional MLKL demonstrated a preserved ability to regenerate, a heightened capacity to produce healthy immune cells, a reduction in DNA damage, and a maintenance of robust mitochondrial function. These beneficial effects were evident even in aged animals and persisted under conditions of imposed stress.
Remarkably, these observed improvements in stem cell function occurred without substantial alterations in global gene expression patterns or chromatin accessibility. This observation strongly suggests that MLKL exerts its pro-aging effects through mechanisms that operate downstream of gene regulation, particularly at the level of cellular ultrastructure, such as the mitochondria, rather than by directly impacting DNA regulation or triggering inflammatory cascades.
The implications of these findings are far-reaching, pointing towards a unifying molecular pathway that bridges diverse cellular stressors to mitochondrial dysfunction and the subsequent aging of stem cells. By pinpointing MLKL as a pivotal nexus in this cascade, the study offers a novel conceptual framework for understanding how aging compromises the integrity and function of the hematopoietic system.
Dr. Yamashita underscored the long-term potential of this research, stating, "In the longer term, this research could lead to therapies that preserve the function of hematopoietic stem cells, ultimately improving recovery and long-term health for patients undergoing chemotherapy, radiation, or transplantation. By revealing how non-lethal activation of cell-death pathways drives stem cell aging, these findings may inspire new classes of mitochondrial-protective or necroptosis-modulating drugs." This perspective highlights the translational promise of the discovery, suggesting potential therapeutic avenues for enhancing resilience and recovery in individuals undergoing treatments that often deplete stem cell reserves.
In summation, this comprehensive study elucidates a previously unappreciated role for MLKL in stem cell aging, demonstrating its capacity to induce functional decline without necessarily triggering cell death. Instead, MLKL acts as a stress-responsive effector that initiates mitochondrial damage, progressively weakening HSC function over time. This groundbreaking discovery challenges conventional understanding of proteins traditionally linked to necroptosis and opens exciting new avenues for the development of interventions aimed at mitigating or preventing age-related deterioration of the blood and immune systems.
About the Author






{kind=link}