
A groundbreaking investigation, recently bolstered by significant funding from the National Institutes of Health, is paving the way for an innovative therapeutic strategy against Alzheimer’s disease, centering on a subtle yet crucial gas produced within the brain. This gas, derived from a protein known for its distinct, albeit pungent, odor reminiscent of sulfurous compounds, is emerging as a critical factor in the intricate mechanisms of memory formation and cognitive health. The findings, meticulously detailed in the esteemed journal Proceedings of the National Academy of Sciences, stem from comprehensive studies conducted on genetically modified rodent models, spearheaded by Dr. Bindu Paul, an associate professor of pharmacology, psychiatry, and neuroscience at Johns Hopkins University School of Medicine.
The protein in question, identified as Cystathionine $gamma$-lyase (CSE), is primarily recognized for its enzymatic function in generating hydrogen sulfide. While commonly associated with an unpleasant smell, this gas, in minute quantities, appears to exert profound protective effects on neuronal function. The current research endeavors to unravel the precise operational pathways of CSE and to ascertain whether enhancing its activity could serve as a viable method to fortify brain cells and decelerate the progression of debilitating neurodegenerative conditions, such as Alzheimer’s disease.
Previous experimental evidence had posited that hydrogen sulfide possesses the capacity to safeguard neurons in laboratory animals. However, the inherent toxicity of hydrogen sulfide in higher concentrations presents a significant hurdle for direct therapeutic administration to the brain. Consequently, the scientific community is now focused on understanding how to maintain the naturally occurring, extremely low concentrations of this gas within neurons in a safe and controlled manner.
The latest findings reveal a compelling correlation between the absence of the CSE enzyme and the manifestation of cognitive deficits. Mice engineered to lack CSE exhibited notable impairments in memory and learning capabilities. Furthermore, these genetically altered subjects displayed elevated markers of oxidative stress, evidence of DNA damage, and a compromised integrity of the blood-brain barrier – a confluence of pathological indicators frequently observed in individuals afflicted with Alzheimer’s disease, as elucidated by Dr. Paul, the study’s corresponding author.
This contemporary research builds upon a rich legacy of prior investigations, notably those led by the distinguished Dr. Solomon Snyder, professor emeritus of neuroscience, pharmacology, and psychiatry. As far back as 2014, Dr. Snyder’s research team had published findings indicating that CSE played a supportive role in brain health within mouse models exhibiting Huntington’s disease. These earlier experiments utilized mice lacking the CSE protein, a strain that had been initially developed in 2008 when the protein’s involvement in vascular function and blood pressure regulation first came to light.
Subsequent to these foundational studies, in 2021, Dr. Snyder’s group observed a dysregulation in CSE functionality in mice afflicted with Alzheimer’s disease. Their research at that time also demonstrated that even minuscule quantities administered via injection of hydrogen sulfide could confer protection to brain function. These earlier investigations primarily focused on mice that possessed additional genetic mutations predisposing them to neurodegenerative disorders. The most recent study, however, has succeeded in isolating and clarifying the specific role of CSE itself, independent of other confounding genetic factors.
"This most recent work unequivocally indicates that CSE, acting on its own, is a pivotal determinant of cognitive function and presents a novel therapeutic avenue for addressing Alzheimer’s disease," stated Dr. Snyder, a co-corresponding author who concluded his tenure at Johns Hopkins Medicine in 2023.
To meticulously examine the impact of CSE on memory processes, the research team conducted comparative analyses between mice devoid of the protein and their normal counterparts, utilizing the same genetically modified strain established in 2008. A key component of their assessment involved evaluating spatial memory – the capacity to recall directional information and navigate environments – employing a behavioral test known as the Barnes maze. In this experimental paradigm, rodents are tasked with locating a concealed shelter to escape a noxious stimulus, typically a bright light.
The results of the Barnes maze test revealed a striking divergence in cognitive performance over time. At the two-month mark, both the control group and the CSE-deficient mice demonstrated comparable abilities in finding the shelter, typically within a three-minute timeframe. However, by six months of age, a significant deterioration in performance was evident among the CSE-deficient mice, which struggled to locate the escape route, while the normal mice continued to exhibit proficient navigation.
"This progressive decline in spatial memory strongly suggests the onset of a neurodegenerative process directly attributable to the loss of CSE," commented Dr. Suwarna Chakraborty, the study’s lead author and a researcher within Dr. Paul’s laboratory.
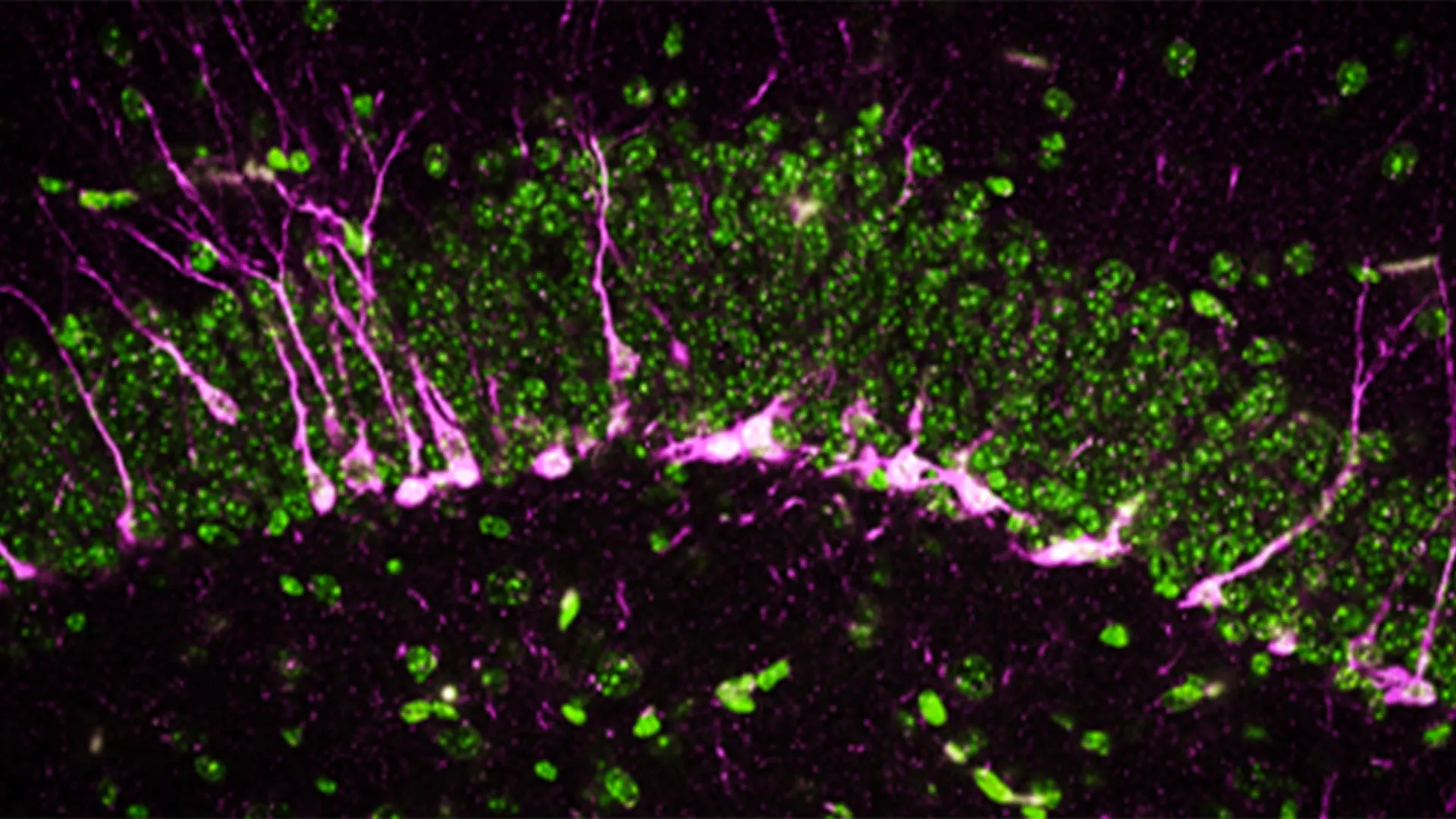
Beyond behavioral observations, the researchers delved into the cellular repercussions of CSE deficiency within the brain. The hippocampus, a brain region indispensable for learning and memory consolidation, is characterized by a continuous process of neurogenesis, the formation of new neurons. Impairments in this generative capacity are a well-established hallmark of neurodegenerative diseases. Through sophisticated biochemical and analytical techniques, the team discovered that proteins crucial for neurogenesis were either significantly diminished or entirely absent in the brains of mice lacking CSE.
Employing high-resolution electron microscopy, the scientists observed profound structural damage within the brains of these CSE-deficient mice. Specifically, they identified substantial disruptions in the integrity of cerebral blood vessels, indicative of damage to the blood-brain barrier – another critical pathological feature associated with Alzheimer’s disease. Furthermore, the study revealed that newly generated neurons encountered considerable difficulties in migrating to the hippocampus, where their integration is essential for effective memory formation.
"The mice lacking CSE exhibited functional deficits at multiple biological levels, mirroring the symptomatic presentation of Alzheimer’s disease," noted Dr. Sunil Jamuna Tripathi, a co-first author and fellow researcher in Dr. Paul’s lab.
Alzheimer’s disease represents a formidable public health challenge, affecting over six million individuals in the United States alone, with projections indicating a continued upward trend in prevalence according to the Centers for Disease Control and Prevention. Critically, current therapeutic interventions have not yet demonstrated a consistent ability to halt or meaningfully slow the inexorable progression of this devastating illness.
The researchers are optimistic that by targeting CSE and modulating its production of hydrogen sulfide, a novel therapeutic paradigm for Alzheimer’s disease may be realized. Such an approach could offer a promising new avenue for developing treatments designed to preserve cognitive function and decelerate disease advancement.
This extensive research was made possible through substantial financial contributions from a diverse array of funding bodies, including the National Institutes of Health (grant numbers 1R01AG071512, P50 DA044123, 1R01AG073684, O1AGs066707, U01 AG073323, AG077396, NS101967, NS133688, P01CA236778), the Department of Defense (grant number HT94252310443), the American Heart Association, the AHA-Allen Initiative in Brain Health and Cognitive Impairment, the Solve ME/CFS Initiative, a Catalyst Award from Johns Hopkins University, the Valour Foundation, the Wick Foundation, a Department of Veterans Affairs Merit Award (I01BX005976), the Louis Stokes Cleveland Department of Medical Affairs Veterans Center, the Mary Alice Smith Funds for Neuropsychiatry Research, the Lincoln Neurotherapeutics Research Fund, the Gordon and Evie Safran Neuropsychiatry Fund, and the Leonard Krieger Fund of the Cleveland Foundation.
The collaborative effort involved numerous distinguished scientists. Beyond Dr. Paul, Dr. Snyder, Dr. Chakraborty, and Dr. Tripathi, key contributors included Richa Tyagi and Benjamin Orsburn from Johns Hopkins University; Edwin Vázquez-Rosa, Kalyani Chaubey, Hisashi Fujioka, Emiko Miller, and Andrew Pieper from Case Western University; Thibaut Vignane and Milos Filipovic from the Leibniz Institute for Analytical Sciences in Germany; Sudarshana Sharma from Hollings Cancer Center; Bobby Thomas from Darby Children’s Research Institute and the Medical University of South Carolina; and Zachary Weil and Randy Nelson from the West Virginia University School of Medicine.
About the Author









{kind=link}