
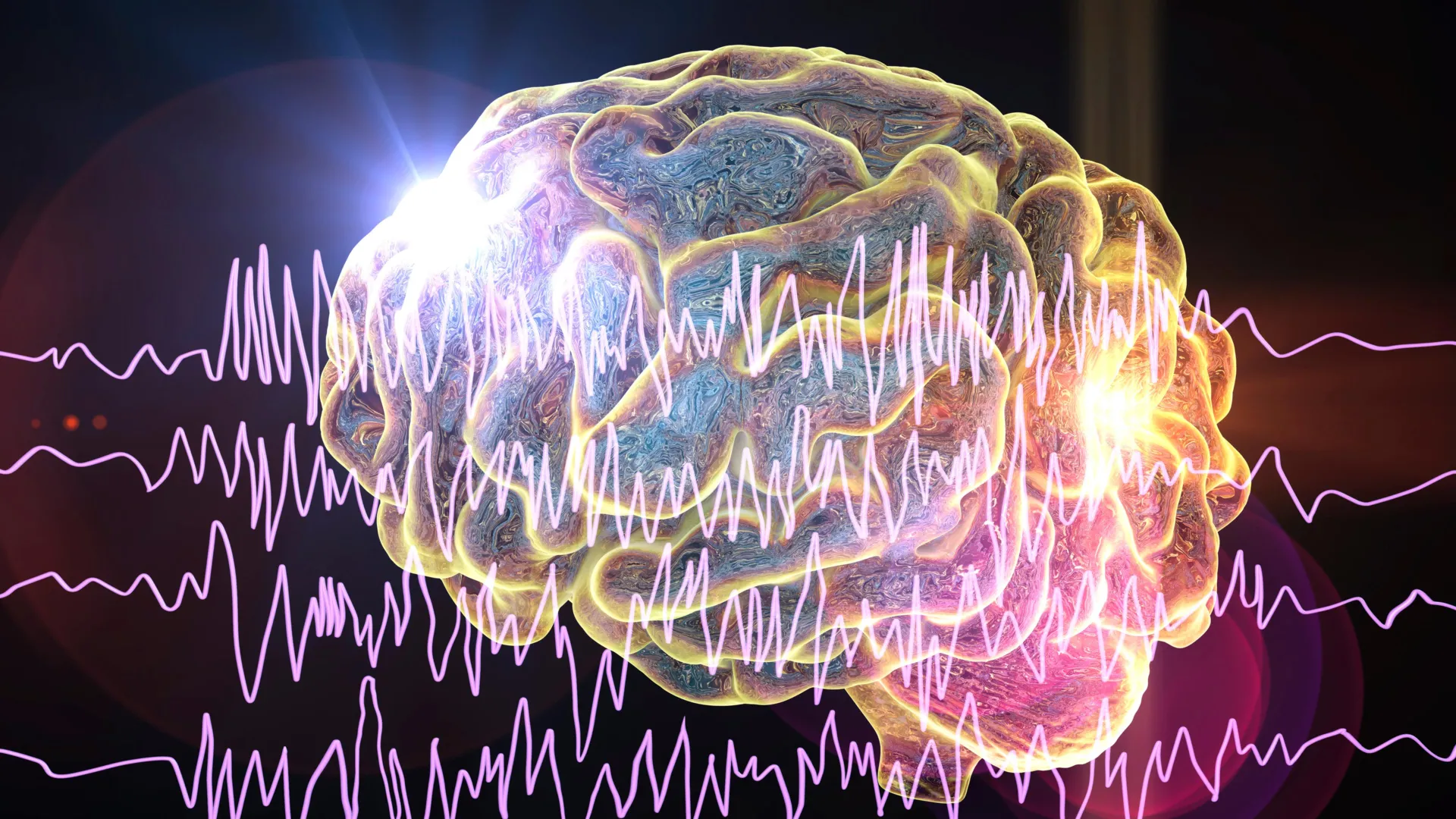
An innovative therapeutic approach, developed through international collaboration and spearheaded by researchers at University College London (UCL) and Great Ormond Street Hospital (GOSH), has demonstrated profound success in significantly curtailing seizure activity in young patients afflicted with a particularly challenging and rare form of epilepsy. Preliminary findings from a comprehensive clinical investigation suggest this novel treatment holds the potential to dramatically enhance the well-being and daily functioning of children grappling with this debilitating condition. The promising outcomes of this experimental therapy, detailed in a recent publication within The New England Journal of Medicine, indicate a potential paradigm shift in managing a disorder that has historically presented formidable treatment obstacles.
The focal point of this research is zorevunersen, an investigational drug that targets the underlying genetic anomaly responsible for Dravet syndrome. In the rigorous clinical trial, children diagnosed with this severe epilepsy experienced a dramatic reduction in seizure frequency, with some participants witnessing a decrease of as much as 91 percent during regular administration of the therapy. Beyond the profound impact on seizure control, early indicators from the study also point towards potential benefits in mitigating some of the cognitive and behavioral deficits commonly associated with Dravet syndrome. Over a three-year observation period, participants exhibited improvements in their overall quality of life, with the majority of reported adverse effects being characterized as mild, suggesting a favorable safety profile.
Dravet syndrome represents a severe, genetic epilepsy characterized by persistent and often intractable seizures, posing a significant therapeutic challenge. This complex neurological disorder is intrinsically linked to a spectrum of long-term neurodevelopmental impairments, including difficulties with cognitive processing, behavioral regulation, feeding, and motor coordination. Tragically, the condition is also associated with an elevated risk of premature mortality, underscoring the urgent need for more effective interventions. The therapeutic landscape for Dravet syndrome has historically been constrained, with existing antiepileptic medications frequently proving insufficient in achieving comprehensive seizure control. Furthermore, no currently approved treatments directly address the pervasive cognitive and behavioral sequelae that significantly impact the lives of affected children and their families.
The innovative mechanism of zorevunersen lies in its targeted approach to the genetic root of Dravet syndrome. The drug, a product of collaboration between Stoke Therapeutics and Biogen, is engineered to rectify the specific genetic defect that underpins the disorder. In the general population, individuals possess two functional copies of the SCN1A gene, which plays a crucial role in neuronal function. However, in individuals with Dravet syndrome, one copy of this gene is compromised, leading to an insufficient production of a vital protein essential for proper nerve cell communication. Zorevunersen functions by stimulating the production of this critical protein from the healthy copy of the SCN1A gene. By augmenting the levels of this protein, the therapy aims to restore more normative electrical signaling within nerve cells, thereby ameliorating the underlying pathophysiology of the epilepsy.
The data emerging from this study comprises findings from both the initial clinical trial and subsequent follow-up extension studies, collectively involving 81 children diagnosed with Dravet syndrome across the United Kingdom and the United States. These early-stage investigations were primarily designed to rigorously evaluate the safety and tolerability of zorevunersen, while simultaneously monitoring its impact on seizure frequency, cognitive function, behavioral patterns, and overall quality of life. The promising results from these foundational trials have paved the way for a larger, pivotal Phase Three study, which is currently underway to further validate the drug’s efficacy and safety in a broader patient population.
Professor Helen Cross, a leading figure in pediatric epilepsy research, serving as Director and Professor of Childhood Epilepsy at the UCL Institute of Child Health and an Honorary Consultant in Pediatric Neurology at Great Ormond Street Hospital, commented on the significance of these findings. She articulated the profound impact of difficult-to-treat genetic epilepsies that extend beyond seizure activity, highlighting the emotional toll on families when treatment options are scarce. Professor Cross expressed optimism that this new therapeutic avenue could offer affected children a pathway to significantly improved health and happiness. She further affirmed that the data collectively indicate zorevunersen to be safe and well-tolerated by the majority of patients, providing a strong rationale for its continued evaluation in the ongoing Phase Three trials.
The initial clinical trial enrolled a cohort of 81 children, aged between two and 18 years, all of whom had previously experienced a substantial burden of seizures, averaging approximately 17 per month before commencing the study. Participants received varying doses of zorevunersen, administered via lumbar puncture, with some receiving a single dose and others undergoing additional administrations at two- to three-month intervals over a six-month treatment period. A significant majority, 75 of the initial participants, opted to continue into the extension studies, where they received the medication at a four-month interval. Analysis of the data revealed a remarkable reduction in seizure frequency among those who received the 70mg dose during the initial phase. Over the first 20 months of the extension studies, these children experienced a decrease in seizure frequency ranging from 59 percent to an impressive 91 percent, when compared to their pre-treatment baseline.
The clinical trial involved a network of leading pediatric hospitals across the United Kingdom. Nineteen of the participants received treatment at UK-based centers, including Great Ormond Street Hospital, Sheffield Children’s Hospital, Evelina London Children’s Hospital, and The Royal Hospital for Children in Glasgow. At Great Ormond Street Hospital, the research was conducted within the National Institute for Health and Care Research’s Clinical Research Facility, a specialized unit dedicated to the advancement of experimental pediatric clinical trials.
Galia Wilson, Chair of Trustees for Dravet Syndrome UK, underscored the profound and often devastating impact of the condition on families. She conveyed immense enthusiasm regarding the recent findings from the initial zorevunersen trials, emphasizing the hope these results may bring. Ms. Wilson expressed keen anticipation for the forthcoming Phase Three clinical trials, viewing them as a crucial step in determining whether the early promise of zorevunersen will translate into tangible hope and improved outcomes for all families affected by Dravet syndrome.
The transformative impact of the therapy was powerfully illustrated by the personal experience of Freddie, an eight-year-old boy from Huddersfield under the care of Sheffield Children’s NHS Foundation Trust, who participated in the trial. Prior to commencing the treatment in 2021, Freddie endured more than a dozen nocturnal seizures each month. Following the initiation of zorevunersen, his seizure pattern underwent a radical alteration, with his seizures diminishing to one or two brief, seconds-long episodes occurring only every three to five days. Freddie’s mother, Lauren, described the trial as life-altering, stating that it had gifted them a life they had previously thought unattainable, and most importantly, a life that Freddie could genuinely enjoy. This anecdotal evidence provides a poignant testament to the potential of zorevunersen to fundamentally improve the quality of life for children living with Dravet syndrome.
About the Author









{kind=link}